VITERBO – Un nuovo studio pubblicato da ricercatori dell’Universita degli Studi della Tuscia getta luce sui complessi meccanismi che governano il movimento dei rotassani, sofisticate strutture molecolari considerate tra i candidati più promettenti per lo sviluppo di macchine molecolari e dispositivi nanotecnologici del futuro.
Lo studio
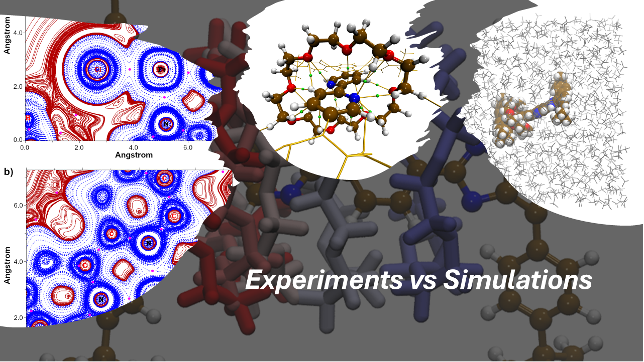
I ricercatori hanno condotto uno studio computazionale approfondito su un rotassano, una struttura in cui un anello molecolare (macrociclo 24-crown-8) è meccanicamente intrecciato su un “filo” molecolare contenente due stazioni simmetriche di benzimidazolo legate a un nucleo centrale di 2,2′-bipiridile.Utilizzando simulazioni avanzate di dinamica molecolare e calcoli basati sulla teoria del funzionale della densità (DFT), il team ha riprodotto quantitativamente le preferenze conformazionali sperimentalmente osservate, caratterizzando il panorama energetico che guida il movimento reversibile dell’anello lungo il filo molecolare a temperatura ambiente in soluzione di diclorometano.
Controllo del movimento molecolare
Un risultato particolarmente significativo dello studio riguarda la capacità di “bloccare” il movimento translazionale naturale del macrociclo tra le stazioni presenti. I ricercatori hanno dimostrato che, in presenza di cloruro di platino (PtCl₂) in soluzione di dimetilformammide (DMF), il complesso assume una forma traslazionalmente inattiva grazie al coordinamento del platino al sito chelante della bipiridile.Nello specifico, la struttura assunta in presenza di platino è stata caratterizzata in dettaglio attraverso descrittori della teoria quantistica degli atomi nelle molecole (QTAIM), fornendo una comprensione molecolare approfondita delle interazioni che stabilizzano il sistema supramolecolare nella forma non responsiva al movimento su nanoscala.
Implicazioni e prospettive“I rotassani rappresentano una classe fondamentale di molecole per lo sviluppo di interruttori molecolari, sensori e dispositivi di stoccaggio dell’informazione su scala nanometrica. La capacità di comprendere e prevedere il comportamento di queste “macchine molecolari” attraverso la modellistica teorica rappresenta un passo importante verso la progettazione razionale di sistemi molecolari funzionali sempre più sofisticati.” riporta il dott. Costantino Zazza ricercatore del Dipartimento DIBAF in Unitus.
“Piattaforme abilitanti al supercalcolo offrono ambienti in cui sperimentare algoritmi sofisticati o proporne delle alternative da associare allo studio degli effetti in natura derivanti da interazioni fondamentali in sistemi flessibili e di grande complessità” conclude in dott. Zazza. Questo studio dimostra come l’approccio computazionale multi-scala, che combina dinamica molecolare classica e calcoli quanto meccanici, possa fornire un quadro dettagliato dei meccanismi che governano il funzionamento di architetture molecolari complesse, accelerando lo sviluppo di future applicazioni nanotecnologiche.